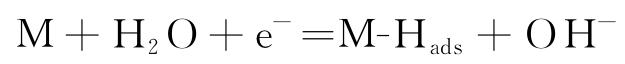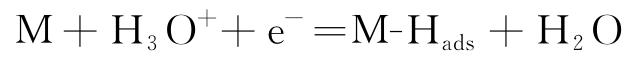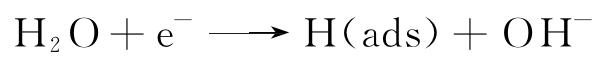表 1 两种600合金试样的化学成分
Table 1. Chemical compositions of two 600 alloy samples
| 试样 | 质量分数/% | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ni | Cr | Fe | C | Mn | Si | Cu | P | S | B | |
| 轧制退火 | 74 | 16.4 | 8.5 | 0.06 | 0.23 | 0.22 | 0.01 | 0.004 | 0.001 | - |
| 固溶退火 | 75.1 | 15.6 | 7.92 | 0.01 | 0.46 | 0.22 | <0.01 | 0.009 2 | - | 0.002 3 |

600合金是一种镍基合金,由于其具有优异的力学性能和耐腐蚀性能,被广泛应用于石油、化工和核电领域。国际早期建造的压水反应堆(PWR)中大量使用了600合金。例如,截至2005年,美国大约50%的PWR仍在使用600合金蒸汽发生管[1]。在我国早期的压水堆核电站一回路系统中,也使用了600合金作为一些关键位置的材料[2]。而在压水堆核电站冷却剂中含有一定量的溶解氢,其浓度是影响合金腐蚀特性的关键因素。因此,研究氢对600合金在反应堆环境中耐腐蚀性能的影响,对于维护早期核电设备的运行安全有重要意义,并且也能为研究同类镍基合金的腐蚀行为提供参考。有关氢对镍基合金氧化膜性质、应力腐蚀开裂(SCC)行为和电化学行为的影响目前已有一些报道[3]。HOU等[4]将预充氢和未充氢的600合金试样在288 ℃的含氧水中持续暴露100 h,结果发现预充氢试样腐蚀速率更快,表面氧化膜更厚且存在缺陷。LAI等[5]通过比较三种不同显微组织的600合金在含溶解氢的锂化水中的阳极极化曲线,发现低温退火(927 ℃)合金的溶解速率更快,钝化性更弱。TOMOKAZU等[6]则发现溶解氢含量对阴极极化曲线影响不大,但对于阳极曲线,电流密度随溶解氢氢含量的增加而增大。
笔者通过四种不同的溶液来模拟600合金的工作环境,采用电化学充氢法研究了两种微观结构的600合金长时间暴露在高温高压水中的电化学行为,并比较了不同微观结构合金在各种水化学条件下的耐腐蚀性能。
分别从太平洋西北国家实验室(PNNL)和通用电气全球研究中心(GE-GRC)获取两种不同的600合金。其中,前者的最后热处理状态为927 ℃保温5 h后水冷(以下简称轧制退火试样),而后者的最后热处理状态为1 100 ℃保温30 min后水冷(以下简称固溶退火试样),试样的化学成分如表1所示,试样尺寸均为2 cm×1 cm×0.2 cm。
| 试样 | 质量分数/% | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ni | Cr | Fe | C | Mn | Si | Cu | P | S | B | |
| 轧制退火 | 74 | 16.4 | 8.5 | 0.06 | 0.23 | 0.22 | 0.01 | 0.004 | 0.001 | - |
| 固溶退火 | 75.1 | 15.6 | 7.92 | 0.01 | 0.46 | 0.22 | <0.01 | 0.009 2 | - | 0.002 3 |
分别通过四种溶液来模拟600合金的工作环境:(1)含氧0.01 mol/L NaCl溶液(以下简称含氧NaCl溶液),利用水浴加热法将该溶液加热至70 ℃,然后将其敞口放置于空气中;(2)脱氧纯水(25 ℃时电导率<0.05 μS/cm),工作温度为(300±0.1) ℃;(3)在脱氧纯水中加入1 200 mg/L H3BO3+2.2 mg/L LiOH来模拟压水堆核电站一回路冷却水(以下简称脱氧模拟水),工作温度为(300±0.1) ℃;(4)在脱氧模拟水中注入氢气,使氢质量分数度为1.8 mg/kg,工作温度为(300±0.1) ℃(以下简称氢化脱氧模拟水)。
采用配备循环水化学回路的316L不锈钢高压釜进行高温电化学测试。高压釜的温度和流量由高压计量和背压调节器控制,试验中高压釜的温度调节为(300±0.1) ℃,工作压力为(12±0.1) MPa,控制循环流量为4 L/h。使用Duke Nuclear Chemicals(美国)开发的pH和电导率计算器软件计算溶液的pH和电导率。通过将高纯度氩气、氢气注入装置的玻璃水箱中,分别制备脱氧、氢化脱氧模拟水。将试样置于高压釜中进行为期190 h的暴露试验,试验结束后,在相同环境中,对部分试样施加60 mA/cm2的电流,进行原位电化学充氢,然后测试动电位极化曲线和计时电流图。
电化学测试采用三电极体系,其中经过模拟腐蚀试验的试样为工作电极,铂为对电极,Cu/Cu2O/ZrO2为参比电极。用Gamry Reference 600恒电位仪/恒电流仪/ZRA记录未充氢和充氢试样的开路电位(OCP)、动电位极化曲线(扫描速率为1 mV/s)、电化学阻抗谱(EIS)和计时电流图(阴极电位-0.5 V,持续时间1 h)。在工作电极达到稳定的OCP后,在-0.5 ~1 V的电位范围内记录动电位极化测量值。电化学阻抗谱记录的频率范围为0.01 Hz~100 kHz,扰动电流振幅为10 mV。根据式(1)将参比电极的电位转换为相对于标准氢电极(SHE)的电极电位E[7]。无特殊说明,文中电位均相对于SHE。
|
|
(1) |
式中:T为试验温度,℃。
暴露试验前,试样经砂纸逐级打磨,抛光布和金刚石膏抛光,蒸馏水和乙醇超声清洗,分别使用光学显微镜(NOVA NanoSEM 230型)和电子背散射衍射(EBSD)对试样的金相组织进行观察。经电化学测试后试样表面形貌使用扫描电子显微镜(SEM,FEI Sirion 200型)进行观察。
由图1可见:通过线性截取法确定固溶退火试样的平均晶粒尺寸为250 μm,轧制退火试样的平均晶粒尺寸为65 μm;固溶退火试样的微观结构更加均匀,晶界光滑,无碳化物颗粒,而轧制退火试样的晶界曲折,有较多的碳化物颗粒。如图2所示,两种试样的晶界上都有氧化物沉淀,轧制退火试样中的碳化物颗粒为Cr23C6,在晶界和晶粒内都有分布,而固溶退火试样内几乎不存在碳化物颗粒。对于600合金的微观组织,类似的情况在文献中亦有报道[8],相比于轧制退火试样,固溶退火试样具有更理想的微观结构。
使用塔菲尔线外推法确定腐蚀电流(Jcorr)[9],在极化曲线中,腐蚀电位(Ecorr)是判断材料在腐蚀性介质中耐腐蚀性能的量度[10],更正的腐蚀电位、更低腐蚀电流密度通常表明材料具有良好的电化学性能。在70 ℃含氧NaCl溶液中,试样在电化学预充氢期间以及动电位极化曲线阴极支路的主要阴极反应是氧和氢的还原反应。
由图3可见:在70 ℃含氧NaCl溶液中,未充氢时,固溶退火试样的Ecorr和击穿电位(Et)分别为0.344 V和0.763 V,腐蚀电流密度为0.09 μA/cm2;相比之下,轧制退火试样的电位(Ecorr=0.285 V,Et=0.548 V)更偏向负值,腐蚀电流密度(Jcorr =0.105 μA/cm2)略高于固溶退火试样。二者的阴极支路电流密度没有显著区别,但轧制退火试样的阳极支路电流密度明显大于固溶退火试样。由此可见,未充氢时,微观结构更理想的固溶退火试样在70 ℃含氧NaCl溶液中电化学性能更好。
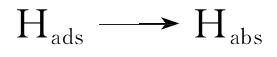
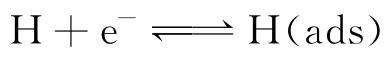
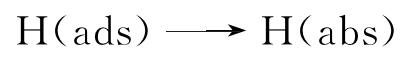
由图3还可见,经过电化学充氢后,两种试样在70 ℃含氧NaCl溶液中的Ecorr和Et都负向偏移,阳极氧化电流密度增加。同时,固溶退火试样的腐蚀电流密度增大至0.11 μA/cm2。这说明经过电化学充氢后,两种试样的耐腐蚀性能均下降。在中性或酸性溶液中进行电化学充氢时,水分子在作为阴极的金属表面发生Volmer反应生成氢原子,吸附在材料表面的氢原子[11]会通过吸收和扩散的方式渗透到材料内部[12],可以用公式(1)~(3)描述这一系列过程。
|
|
(2) |
|
|
(3) |
|
|
(4) |
式中:M表示金属表面。
吸收/渗透的氢原子会使合金表面的耐腐蚀性能下降,尤其是在溶液中存在氯离子的情况下,性能下降更加显著[13]。由图4可见,在70 ℃含氧NaCl溶液中,预充氢时,两种试样表面没有点蚀痕迹,仅在固溶退火试样表面发现腐蚀产物。
使用pH和电导率计算软件,结合水的解离常数和H3O+、OH-的分布变化,可计算在300 ℃下水的pH下降至5.7,同时电导率增加至5.6 μS/cm。
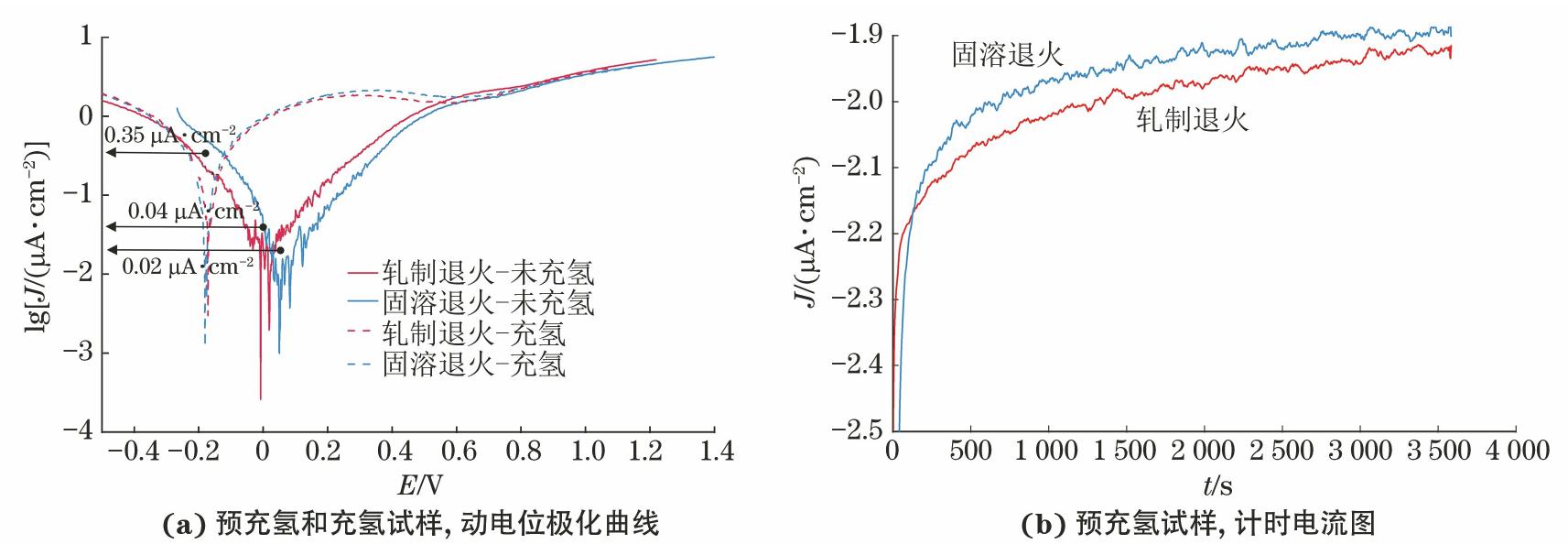
由图5(a)可见:当未充氢时,在高温脱氧纯水中固溶退火试样的腐蚀电流密度为0.02 μA/cm2,小于轧制退火试样的0.04 μA/cm2,且其腐蚀电位更正。这表明未充氢时,固溶退火试样的电化学性能略好于轧制退火试样。然而,进行电化学充氢后,固溶和轧制退火试样的动电位极化曲线几乎重叠,Ecorr向负向偏移至-0.180 V,Jcorr为0.35 μA/cm2,比未充氢试样增大了数十倍,表明其耐腐蚀性能降低。
在190 h暴露试验过程中,试样表面同时发生氧化、溶解、沉积过程,试验结束后形成了稳定的氧化膜[14-16]。在电化学充氢过程中,氢原子在试样表面发生如式(5)~(7)所示的渗透/吸收作用,这会改变氧化膜的性质[14-15,17-19],从而影响材料的电化学行为。本试验中由于两种合金试样具有不同的微观结构,化学成分也有略微差异,会影响形成的氧化膜的结构[20],因此表现出不同的电化学行为,但在氢的作用下,二者耐腐蚀性能均下降。
|
|
(5) |
|
|
(6) |
|
|
(7) |
计时电流图所示的阴极电流密度主要由暴露试验后合金电化学充氢过程中溶解氢的还原产生。由图5(b)可见,在高温脱氧纯水中电化学充氢后,轧制退火试样表面氢的还原反应速率高于固溶退火试样。
根据所使用的计算软件,在300 ℃下脱氧模拟水的pH为6.9,电导率为190 μS/cm。
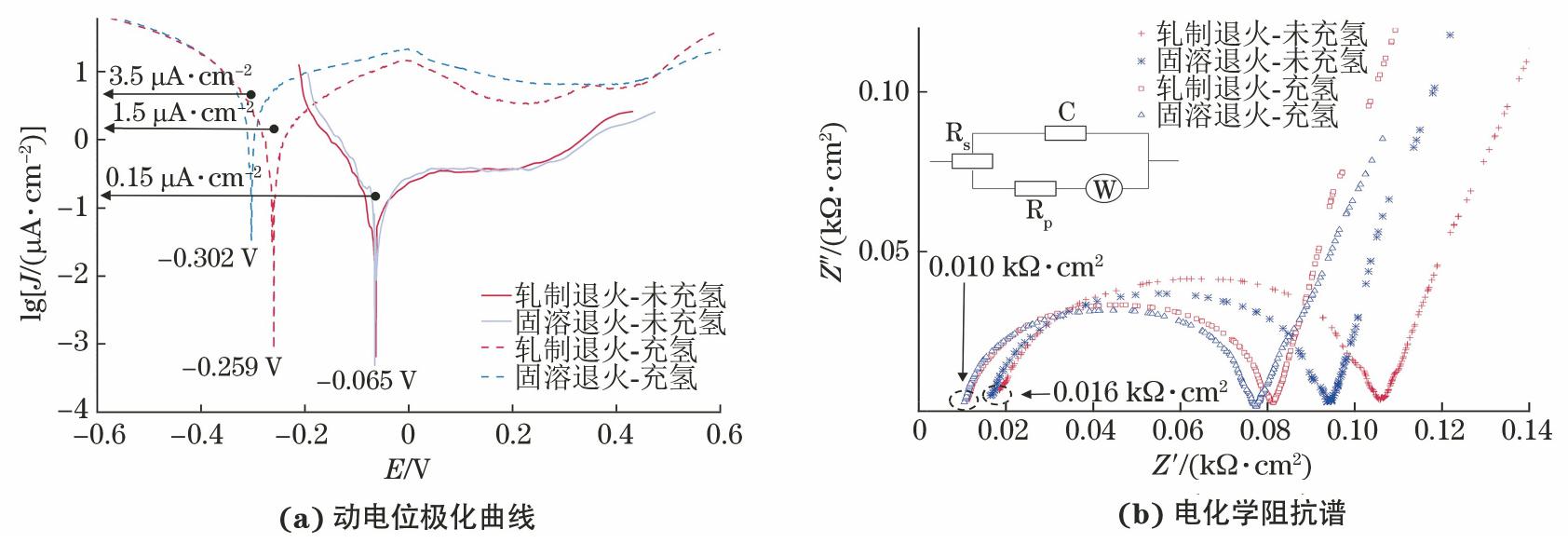
由图6(a)可见:在脱氧模拟水中,两种未充氢试样的动电位极化曲线没有显著差异,这两种合金的Ecorr约为-0.065 V,腐蚀电流密度约为0.15 μA/cm2;电化学充氢后,两种试样的Ecorr都负向偏移(轧制退火试样和固溶退火试样的Ecorr分别为-0.259 V,-0.302 V),腐蚀电流密度和氧化电流密度增大了10倍以上,这说明电化学充氢使试样的耐腐蚀性能降低。
与在脱氧纯水中类似,600合金试样在脱氧模拟水中暴露190 h后,表面也会形成具有双层结构的氧化膜[6,16,18,21-22]。图6(b)所示为4组试样的电化学阻抗谱和拟合的等效电路。其中,Rs为溶液电阻,Rp为氧化层电阻,W为Warburg阻抗。在4组试样中,未充氢轧制退火试样的Rp值最大;在电化学充氢后,电化学阻抗谱的半圆直径减小,两种合金表面氧化层的电阻率降低。在已形成氧化膜的情况下,电化学充氢依然会改变试样的电化学性能。这说明,氢的吸收/渗透作用会改变氧化膜的性质[4,21,23],破坏其稳定性。
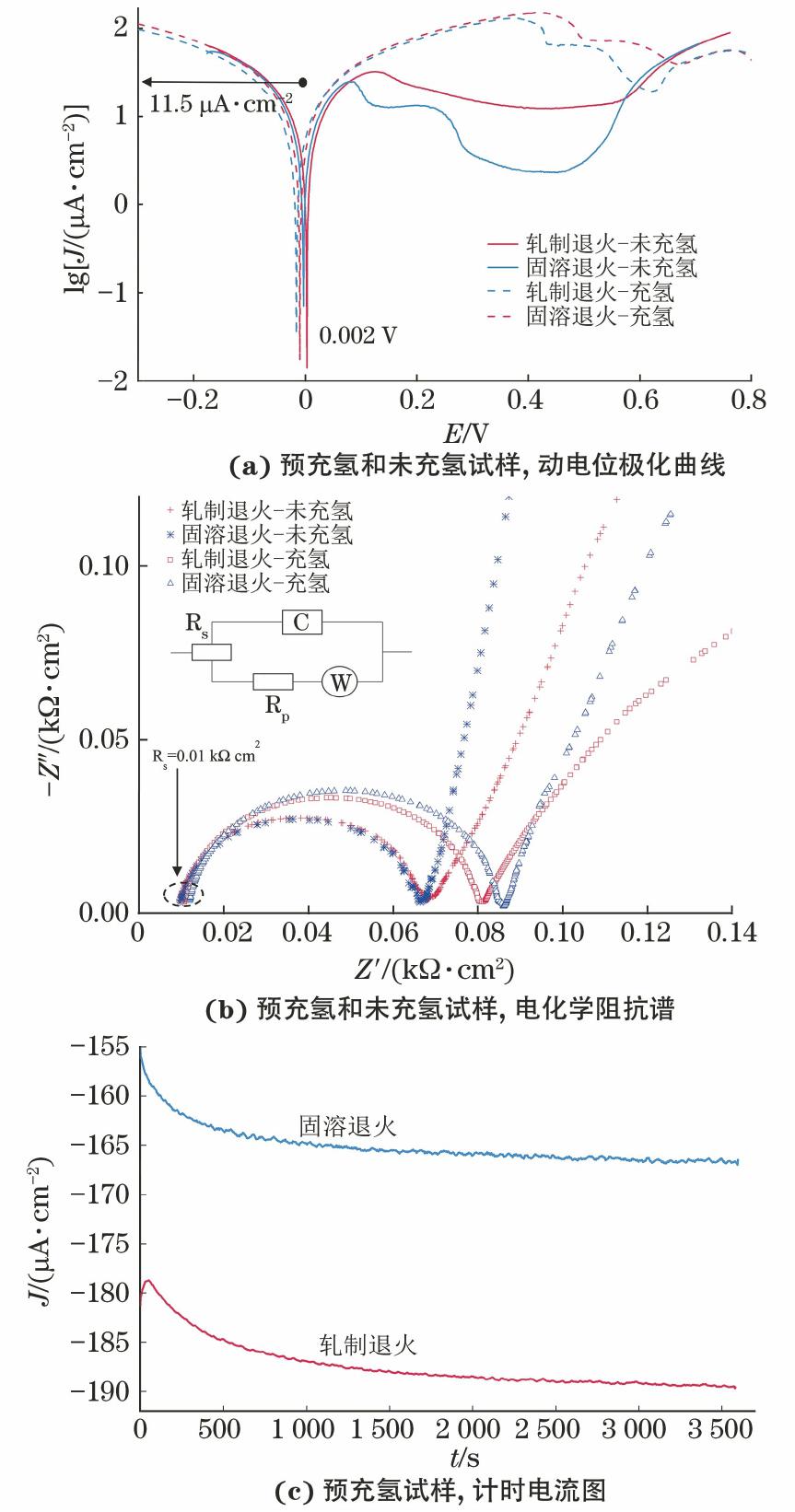
由图7(a)可见,4个试样的Ecorr均为0.002 V,这说明在氢化脱氧模拟水中长时间暴露后,1 h的电化学充氢无法对试样的腐蚀电位产生明显影响。但是,电化学充氢试样的氧化电流密度高于未充氢试样,充氢前后固溶退火试样的氧化电流密度比轧制退火试样更小。
由图7(b)可见,预充氢时,与脱氧模拟水中测得的结果相反,试样在氢化脱氧模拟水中的Rp值增大,说明在氢化脱氧模拟水中进行电化学充氢会使氧化膜的钝化作用增强。此外,充氢后轧制退火试样的阻抗谱低频区域的Warburg阻抗表现出非理想行为,其斜率低于理想的Warburg阻抗斜率(45°)。Warburg阻抗的斜率是分析Nyquist图的一个重要参数,它与表面的离子电阻直接相关。当斜率减小时,表面的离子电阻增加[24]。根据文献[6,15-16]报道,试样暴露在含溶解氢的环境中所生成氧化膜的厚度与氢含量成反比,并且氧化膜会出现孔隙。而将氧化后的试样进行原位充氢,则能观察到更明显的“空穴”,氧化膜呈现多孔性[14-15,17]。研究表明,在图7(b)曲线图中观察到的非理想行为是表面粗糙度或孔隙度、现有电容器中存在泄漏或电流分布不均匀等因素造成的[25]。因此,在本试验中观察到Warburg阻抗的非理想行为与充氢后氧化膜结构的改变有关。
由图7(c)可见,在氢化脱氧模拟水中,预充氢后,轧制退火试样的还原反应速率远高于固溶退火试样,这一结果与在脱氧模拟水中测得的结果一致。
(1)在70 ℃ NaCl溶液、300 ℃脱氧纯水、300 ℃脱氧模拟水中,电化学充氢使两种合金试样的腐蚀电位负向移动,在氢化脱氧模拟水中,合金动电位极化曲线的氧化电流密度均增大,表明在这些环境中电化学充氢使两种合金试样的耐蚀性降低。
(2)在NaCl溶液、脱氧纯水和氢化脱氧模拟水中,固溶退火合金试样表现出更理想的耐蚀性。
(3)在脱氧纯水和氢化脱氧模拟水中,轧制退火合金试样表面的氢还原速率高于固溶退火合金试样。
文章来源——材料与测试网
浙江国检检测技术股份有限公司 版权所有 【暂无】 百度统计
全国统一服务热线:400-1188-260
客服手机号:13372307781
电话:400 1188 260 质量投诉 +86-573-86161208
邮箱:shhgj@chinazbj.com
地址:浙江省嘉兴市海盐县武原街道丰潭路777号
备案号:浙ICP备05056915号
 浙公网安备 33042402000106号
浙公网安备 33042402000106号
技术支持:追马网
 客服微信号
客服微信号
 微信公众号
微信公众号