新污染物是指具有生物毒性、环境持久性和生物累积性等特征,对生态环境或人体健康存在较大风险,但尚未纳入环境管理,或者现有管理措施不足以应对的有毒有害化学物质。2022年5月,国务院印发了《新污染物治理行动方案》,该方案指出,目前国内外广泛关注的新污染物主要有三大类:一是持久性有机污染物;二是内分泌干扰物;三是抗生素。六溴环十二烷(HBCDs)和四溴双酚A(TBBPA)属于持久性有机污染物,近年来作为新污染物受到广泛关注[1-2]。这两类物质是常见的溴代阻燃剂,因其优良的阻燃性能,被广泛应用于建筑材料、车辆、纺织品和家用电器等产品的生产与加工过程中[3-4]。已有研究证实HBCDs和TBBPA具有生物富集性[5],并且对人体及哺乳动物有一定毒性[6-7]。国内外研究表明,这两类物质可在各类环境介质和生物体中被不同程度地检出[8-9]。为了减少HBCDs带来的污染,我国在2021年底全面禁止HBCDs的生产和使用;2023年生态环境部制定了《重点管控新污染物清单(2023年版)》,明确HBCDs属于已淘汰类新污染物,禁止其生产、加工使用以及进出口。尽管目前尚无明确禁止TBBPA生产和使用的法规,但作为新污染物,其与HBCDs性质相似,也存在污染环境的风险。因此,为加强对这两类物质的风险管控,提高新污染物治理能力,建立针对这两类物质的快速、有效、可靠且经济的监测方法十分必要。
我国在HBCDs和TBBPA的监测技术方面起步较晚,分析方法以液相色谱-串联质谱法[10-11]为主,大多聚焦在食品、土壤和生物体中[12-13],对水体的测定研究较少。水中HBCDs和TBBPA的前处理方法主要是液液萃取法,如海洋行业标准HY/T 261—2018《海水中六溴环十二烷的测定 高效液相色谱-串联质谱法》测定海水中的HBCDs时,使用40 mL正己烷分两次对500 mL海水样品进行液液萃取;文献[10]使用100 mL二氯甲烷分两次液液萃取水中的HBCDs和TBBPA;文献[14]使用100 mL二氯甲烷分两次液液萃取水中的HBCDs,这些方法均需要消耗大量的有机溶剂。同时,大部分监测方法会在样品采集后直接过滤除去水样中的悬浮物[14-17],但并未考虑悬浮物中是否含有HBCDs和TBBPA,造成测定结果偏低。因此,本工作采用固相萃取-液相色谱-串联三重四极杆质谱法同时测定水中3种HBCDs(包括α-HBCD、β-HBCD、γ-HBCD)和TBBPA的含量,将水样中的悬浮物和水样作为一个整体,对样品的前处理和仪器工作条件进行了优化,可满足大批量水样的监测分析要求。
1. 试验部分
1.1 仪器与试剂
1290 UPLC-6460 QQQ MSD型高效液相色谱-串联质谱仪;Auto Vap S8型全自动定量浓缩仪;Visiprep DL24位型固相萃取装置;ChromP固相萃取柱(500 mg/6 mL);HLB固相萃取柱(500 mg/6 mL);GCB固相萃取柱(500 mg/6 mL);氨基柱(500 mg/6 mL)。
3种HBCDs混合标准溶液:α-HBCD、β-HBCD、γ-HBCD的质量浓度均为100.0 mg·L−1,溶剂为甲醇,编号1ST80883-100M。
TBBPA标准溶液:100.0 mg·L−1,溶剂为甲醇,编号1ST8713-100M。
碳同位素标记的α-HBCD(13C12-α-HBCD)标准溶液:50.0 mg·L−1,溶剂为甲苯,编号CLM-7922-S。
碳同位素标记的β-HBCD(13C12-β-HBCD)标准溶液:50.0 mg·L−1,溶剂为甲苯,编号CLM-7923-S。
碳同位素标记的γ-HBCD(13C12-γ-HBCD)标准溶液:50.0 mg·L−1,溶剂为甲苯,编号CLM-7924-S。
碳同位素标记的TBBPA(13C12-TBBPA)标准溶液:50.0 mg·L−1,溶剂为甲醇,编号CLM-4694-S。
氘代同位素标记的α-HBCD(α-HBCD-d18)标准溶液:50.0 mg·L−1,溶剂为甲苯,编号DaHBCD。
混合标准中间液:分别取适量的3种HBCDs混合标准溶液和TBBPA标准溶液,用甲醇稀释并定容,配制成质量浓度为10.0 mg·L−1的混合标准中间液。
提取内标溶液:分别取适量的13C12-α-HBCD标准溶液、13C12-β-HBCD标准溶液、13C12-γ-HBCD标准溶液和13C12-TBBPA标准溶液,用甲醇稀释并定容,配制成质量浓度为1.00 mg·L−1的提取内标溶液。提取内标溶液用于修正样品在前处理过程中产生的误差。
进样内标溶液:取适量的α-HBCD-d18标准溶液,用甲醇稀释并定容,配制成质量浓度为1.00 mg·L−1的进样内标溶液。进样内标溶液用于修正仪器进样过程中产生的误差。
混合标准溶液系列:取适量的混合标准中间液,用甲醇逐级稀释,加入适量的提取内标溶液和进样内标溶液,配制成α-HBCD、β-HBCD、γ-HBCD、TBBPA的质量浓度为2.00,5.00,10.0,20.0,50.0,100,200 μg·L−1,提取内标和进样内标的质量浓度为20.0 μg·L−1的混合标准溶液系列。
甲醇、乙腈均为色谱纯;丙酮、二氯甲烷均为农残级;试验用水为超纯水。
1.2 仪器工作条件
1.2.1 色谱条件
Extend C18色谱柱(100 mm×2.1 mm,3.5 μm);流量0.3 mL·min−1;进样量10.0 μL;柱温35 ℃;流动相A为水,B为体积比1∶1的甲醇-乙腈混合溶液。梯度洗脱程序:0~3 min,B由30%升至80%;3~9 min,B由80%升至93%;9~10 min,B由93%降至30%,保持2 min。
1.2.2 质谱条件
电喷雾离子(ESI)源,负离子(ESI−)模式扫描;干燥气温度320 ℃,干燥气流量6 L·min−1;雾化气压力241.3 kPa;鞘气温度340 ℃,鞘气流量11 L·min−1;毛细管电压1 500 V;喷嘴电压2 000 V;多反应监测(MRM)模式。其他质谱参数见表1。
1.3 试验方法
1.3.1 样品采集与保存
按照HJ 91.1—2019《污水监测技术规范》的相关要求,用棕色采样瓶采集样品,确保满瓶采集。在每升水样中加入80 mg硫代硫酸钠和5 mL甲醇,混匀,于4 ℃以下避光运输、保存,14 d内完成样品前处理,30 d内完成分析。
1.3.2 样品制备
取1 L样品,加入20.0 μL提取内标溶液,混匀,抽滤过0.45 μm滤膜,收集滤液。将抽滤后的滤膜剪碎置于15 mL离心管中,加入5 mL丙酮,超声提取20 min,过0.45 μm聚四氟乙烯(PTFE)针头式过滤器,该滤液与抽滤后的滤液合并,过ChromP固相萃取柱(依次用二氯甲烷、丙酮、甲醇和水各10 mL活化),氮气吹扫固相萃取柱10 min,用15.0 mL体积比9∶1的二氯甲烷-丙酮混合溶液洗脱,收集洗脱液置于浓缩仪中,氮吹至近干,残余物用甲醇溶解并定容至1.0 mL,加入20.0 μL进样内标溶液,混匀,过0.22 μm PTFE针头式过滤器,按照仪器工作条件测定。
2. 结果与讨论
2.1 前处理方法的优化
2.1.1 固相萃取柱
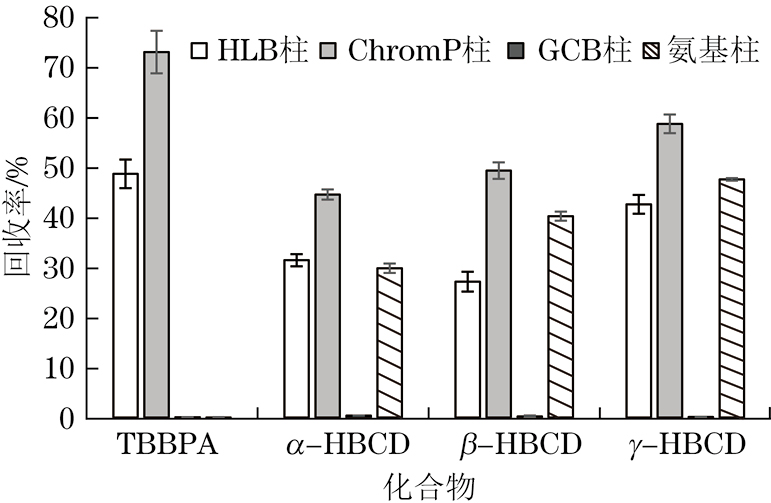
固相萃取法具有操作简单、溶剂消耗少,能同时完成萃取和净化,并可实现自动化操作等优势。常用于样品萃取和净化的固相萃取柱有键合硅胶固相萃取柱(如氨基柱)、无机固相萃取柱(如GCB柱)、高聚物固相萃取柱(如HLB柱、ChromP柱),试验考察了上述4种固相萃取柱对各目标化合物萃取效果的影响,结果见图1。
由图1可知:采用GCB柱时,HBCDs和TBBPA回收率基本为0,可能由于GCB柱对含有苯环官能团的目标化合物(如TBBPA)具有较强的吸附能力[12,18],HBCDs虽不含苯环但与TBBPA结构相似,导致这两类目标化合物难以被洗脱;采用氨基柱时,TBBPA回收率基本为0,可能由于TBBPA存在两个酚羟基,属于离子型有机污染物,其pKa1为7.5,在中性环境中,TBBPA部分会以阴离子的形式存在,且TBBPA的极性大于HBCDs的,导致其在氨基柱上的保留能力过强,难以被洗脱;采用HLB柱和ChromP柱时,4种目标化合物均能被萃取,可能由于HLB柱和ChromP柱填料均为亲水亲脂型聚合物,可萃取的化合物极性范围较宽,而采用ChromP柱的回收率均高于HLB柱的,可能因为ChromP柱的填料是苯乙烯-二乙烯基苯,更适合萃取芳香型化合物[19-20]。因此,试验选择的固相萃取柱为ChromP柱。
2.1.2 洗脱溶剂及用量
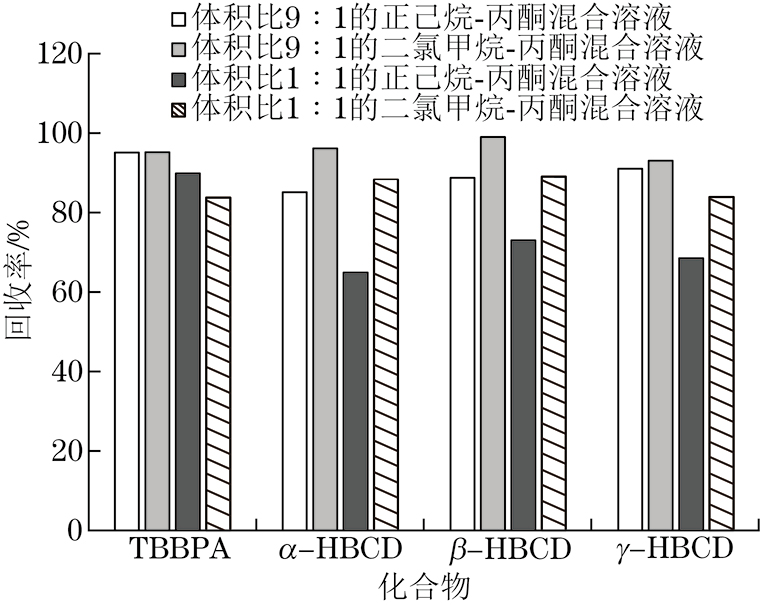
试验首先尝试以甲醇为洗脱溶剂,考察了甲醇用量对各目标化合物回收率的影响。结果显示:当甲醇用量为30 mL时,TBBPA的回收率为80.0%,但HBCDs的回收率仅为50.0%;继续增大甲醇用量到50 mL时,HBCDs的回收率仍只有60.0%,可能由于HBCDs是非极性化合物,甲醇的洗脱能力不足。因此,试验选择洗脱能力更大的溶剂,包括丙酮、二氯甲烷、正己烷及其混合溶液,考察了不同洗脱溶剂(体积比9∶1的正己烷-丙酮混合溶液、体积比1∶1的正己烷-丙酮混合溶液、体积比9∶1的二氯甲烷-丙酮混合溶液、体积比1∶1的二氯甲烷-丙酮混合溶液)对4种目标化合物洗脱效果的影响,结果见图2。
由图2可知:采用体积比1∶1的正己烷-丙酮混合溶液作为洗脱溶剂时,3种HBCDs的回收率较差;采用其余3种混合溶液作为洗脱溶剂时,4种目标化合物的回收率均大于80.0%。考虑到二氯甲烷的挥发性优于正己烷的,且采用体积比9∶1的二氯甲烷-丙酮混合溶液作为洗脱溶剂时,4种目标化合物的回收率最高,试验选择的洗脱溶剂为体积比9∶1的二氯甲烷-丙酮混合溶液。
试验进一步考察了不同用量(1.5,3.0,4.5,6.0,7.5,9.0,10.5,12.0,13.5,15.0 mL)洗脱溶剂对4种目标化合物回收率的影响。结果显示,当洗脱溶剂用量为10.5 mL时,4种目标化合物的回收率基本达到最大,继续增大洗脱溶剂用量并不能提高目标化合物的回收率。考虑到不同批次固相萃取柱的差异,试验选择的洗脱溶剂用量为15.0 mL。
2.1.3 溶液酸度
由于TBBPA存在两个酚羟基,溶液酸度会影响TBBPA的存在状态,试验以空白加标样品(加标量为5.0 ng·L−1)为研究对象,考察了不同溶液酸度(pH 1,2,4,7,8)对4种目标化合物回收率的影响。结果显示:当溶液pH为1时,4种目标化合物的回收率均较低;当溶液pH为2~8时,4种目标化合物的回收率没有明显差异,可能与ChromP柱的填料为亲水亲脂型聚合物有关。为了简化操作,提高样品前处理效率,试验不额外调节溶液酸度。
2.1.4 水样中悬浮物的前处理
试验以加标地表水(加标量为20 ng·L−1)为研究对象,分别对经0.45 μm滤膜抽滤后的水样、抽滤后的滤膜(水样中悬浮物以颗粒物形式吸附在滤膜上)进行前处理,考察了4种目标化合物在水中和颗粒物中的分布状况。结果显示,TBBPA在抽滤后的滤膜上基本未被检出,而HBCDs有近33%(质量分数)分布在颗粒物中,可能与目标化合物的性质有关,HBCDs是非极性化合物,而TBBPA具有一定极性且存在两个酚羟基,相较于HBCDs,TBBPA在水中具有更好的溶解性。因此,测定水样时需对抽滤后的滤膜进行提取。
超声是固体样品常用的提取方法,利用“空化效应”产生的冲击波,使溶液湍流加快,加速热传递并与溶质分散,从而增强提取溶剂与水样间的质量传输,提高提取效率。试验固定超声提取时间20 min,提取溶剂5 mL,考察了不同提取溶剂(甲醇、丙酮和二氯甲烷)对抽滤后的滤膜中HBCDs提取效率的影响。结果显示,采用丙酮和二氯甲烷提取时,HBCDs的回收率相差不大,但采用甲醇提取时,回收率较低,这可能与HBCDs的非极性性质有关。考虑到超声提取液中可能含有基质干扰物质,需对超声提取液进行固相萃取净化处理,而二氯甲烷与水不互溶,试验选择丙酮作为提取溶剂。
2.2 色谱条件的优化
研究表明,流动相中乙腈有利于β-HBCD和γ-HBCD的分离,甲醇有利于α-HBCD和β-HBCD的分离[21]。试验考察了流动相体系中的不同有机相(甲醇、乙腈、体积比1∶1的甲醇-乙腈混合溶液)对3种HBCDs分离效果的影响。结果显示:当有机相为甲醇时,β-HBCD和γ-HBCD色谱峰的分离度较低;当有机相为乙腈时,α-HBCD和β-HBCD色谱峰的分离度较低;当有机相为体积比1∶1的甲醇-乙腈混合溶液时,3种HBCDs色谱峰的分离效果最好,且与基线分离。因此,试验选择体积比1∶1的甲醇-乙腈混合溶液作为流动相体系中的有机相。
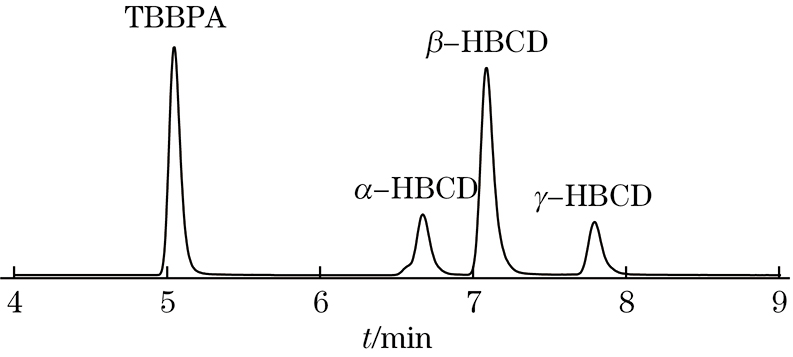
增大进样量能提高方法的灵敏度,但进样量过大可能影响目标化合物的分离度和峰形,进而影响目标化合物的定性定量。试验以100 μg·L−1混合标准溶液为研究对象,考察了进样量为1.0~20.0 μL时4种目标化合物峰形的变化情况。结果显示:当进样量由1.0 μL增大至10.0 μL时,4种目标化合物的峰形无明显变化,峰响应强度则逐渐增强;当进样量增大至20.0 μL时,4种目标化合物均出现不同程度的前展现象,其中TBBPA的峰形变化最为明显。这可能因为混合标准溶液的溶剂为甲醇,而初始流动相仅含30%(体积分数)有机相,当进样量较小时,目标化合物在色谱柱中的扩散尚可在较短时间内完成,不影响目标化合物的峰形,而随着进样量逐渐增大,目标化合物总量逐渐增大,当进样量增大到一定量时,目标化合物在色谱柱前端无法充分扩散,进而导致峰形发生扭曲。为了保证色谱峰峰形,同时得到最佳的检出限,试验选择的进样量为10.0 μL,得到的MRM色谱图见图3。
2.3 标准曲线、检出限和测定下限
按照仪器工作条件测定混合标准溶液系列,以目标化合物与提取内标的质量浓度之比为横坐标,以目标化合物与提取内标的响应强度之比为纵坐标绘制标准曲线。结果显示,4种目标化合物标准曲线的线性范围均为2.00~200 μg·L−1,线性回归方程、相关系数见表2。
平行配制7份空白加标样品(加标量为2.00 ng·L−1)并进行测定,依据HJ 168—2020《环境监测分析方法标准制订技术导则》计算标准偏差s,以3.143s计算检出限,以4倍检出限计算测定下限,结果见表2。
2.4 精密度和回收试验
对地表水样品进行3个浓度水平的加标回收试验,每个浓度水平重复测定6次,计算回收率和测定值的相对标准偏差(RSD),结果见表3。
由表3可知,4种目标化合物的回收率为88.4%~129%,测定值的RSD为2.3%~15%,表明该方法准确度和精密度较好,可以满足痕量分析的需求。
2.5 方法比对
将本方法与现有的分析方法进行比对,结果见表4。
由表4可知:样品的前处理方法主要为液液萃取或固相萃取。采用固相萃取时,由于水样中含有一定量的悬浮物,一般会使用0.45 μm滤膜对样品进行预处理,而本方法显示悬浮物中存在一定量的HBCDs,因此测定水样时需考虑悬浮物中HBCDs的含量;采用液液萃取时,虽然萃取了悬浮物中的目标化合物,但需要消耗较多的有机溶剂。本方法将水样中的悬浮物和水样作为一个整体,既萃取了水样和悬浮物中的目标化合物,又大幅减少了有机溶剂的消耗,检出限等方面优于部分文献,能满足水样中HBCDs和TBBPA的测定需求。
2.6 样品分析
按照试验方法分析在湖北省内采集的32个地表水样品和2个废水样品。结果显示:在地表水样品中,29个样品中均未检出4种目标化合物,1个样品中检出TBBPA,检出量为1.4 ng·L−1,1个样品中检出3种HBCDs,检出量为0.5~1.7 ng·L−1,1个样品中检出α-HBCD、TBBPA,检出量分别为1.9,1.4 ng·L−1;在废水样品中,2个样品中均检出TBBPA,检出量分别为1.4,17.8 ng·L−1。检测结果与文献[25-26]基本一致,说明湖北省的部分水体已存在轻微的HBCDs和TBBPA污染。
本工作采用固相萃取-液相色谱-串联三重四极杆质谱法同时测定水中3种HBCDs和TBBPA的含量。该方法前处理简便高效、可自动化,灵敏度、精密度和准确度较高,可满足水样中HBCDs和TBBPA的测定。
文章来源——材料与测试网





 浙公网安备 33042402000106号
浙公网安备 33042402000106号

